

One of the key aspects of viral protein research is the study of protein domains that remain mostly conserved across many different virus particles. These protein domains are critical for the virus’ propagation and survival, regulating important functions such as host recognition and genome insertion. One such domain is the ACE2 receptor binding domain, critical to the structure of coronavirus spike protein, which rose to public prominence after the COVID-19 pandemic. Spike proteins assemble into trimers on the surface of the mature virion and are found in all coronaviruses. Spike protein trimers are then used by the virion to recognize a potential host and initiate an infection. The importance of spike protein to coronavirus survival has made it a target for both basic and applied research, the former being interested in studying mechanisms of viral infection, the latter interested in developing vaccines to prevent viral infection. This article will focus primarily on the ways researchers are synthesizing this protein in the lab to facilitate its study.
History of Spike Protein
The first infections caused by coronaviruses affected livestock and were observed in the early 20th century. Coronaviruses would not get their name, however, until the 1960s when viral particles were isolated and visualized using transmission electron microscopy (TEM) [1]. These particles possessed a round capsid and crown-shaped protrusions (“corona” being Latin for crown), which would later become known as spike protein. The spike protein first came to prominence in the early 2000s, when a new disease-causing virus began rapidly spreading through regions of mainland China. This disease would later become known as Severe Acute Respiratory Syndrome (SARS) and would be the first instance of widespread human-infecting coronaviruses. Aside from the aforementioned spike protein, other key coronavirus proteins were subsequently identified, these being the envelope (E), membrane (M), and nucleocapsid (N) proteins [2]. While SARS spread rapidly after its initial emergence, proper quarantine procedures prevented it from becoming a global pandemic. The next coronavirus to emerge caused the Middle Eastern Respiratory Virus (MERS) outbreak in 2012. Spike proteins were subsequently discovered to be heavily glycosylated, which spawned several hypotheses into how this facet of spike protein structure plays a role in its function [3,4]. Structural studies suggest that glycosylation is important not only for the recognition of potential host cells but also for proper folding and stabilization of the spike protein fusion trimers. Much like SARS-CoV, MERS-CoV consists of a protein capsid enclosed by a lipid membrane, with spike protein trimers decorating the surface. The conservation of spike protein structure across many different types of coronaviruses would further cement its importance. Spike protein research would explode in popularity after the outbreak of the COVID-19 pandemic, which spurred interest in all facets of coronavirus structure to develop better therapeutics for these types of viruses [4]. While many other types of coronaviruses have been discovered, both with human and non-human hosts, the main three of interest are SARS-CoV, MERS-CoV, and SARS-CoV2. While slight differences in spike protein structure between these three viruses do exist, the key domains of protein structure are mostly consistent.
Ways to Characterize It
Like all proteins, the function of spike protein is dictated primarily by its structure. Thus, spike protein is characterized by methods that allow researchers to obtain high-resolution structural data about its subunits in isolation and a fully assembled trimer. The predominant technique for studying spike protein structure is cryo-electron microscopy (Cryo-EM). This technique is a modification of traditional transmission electron microscopy (TEM) wherein aqueous samples are embedded onto a carbon-film grid then rapidly cooled. Samples prepared in this manner are protected from degradation by the high-energy electron beam and the vacuums that are associated with TEM, furthermore, the crystalline arrangement of biomolecules in the sample provides a myriad of perspectives that are used to generate a 3D image of the sample [5]. Cryo-EM has been used successfully to obtain structures of individual proteins, however, this becomes increasingly difficult with smaller proteins [5]. To circumvent this size issue, Cryo-EM is primarily used for large proteins or protein complexes such as spike protein trimers. Cryo-EM can also be used for the structure of two different proteins in association, such as a spike protein bound to its receptor. All of this structural data is vital to characterizing spike protein, whether it be the changes in structure between variants of coronaviruses or the key residues that are associated with spike protein binding to human receptors. Another commonly used structural technique is Nuclear Magnetic Resonance (NMR) spectroscopy. In NMR, a sample is exposed to a powerful magnetic field which causes molecular dipoles (the sources of which are typically carbon and hydrogen atoms for protein NMR) to align with this magnetic field. Once the magnetic field dissipates, the dipoles then shift back to their original positions. This shift is then detected by the instrument and then deconvoluted by a series of Fourier transforms to yield a final 2D spectrum [6]. This 2D spectrum is sufficient to determine the structure of small molecules, for proteins, a 3D spectrum is required. To obtain a 3D spectrum, all carbons and nitrogens in the protein must be isotopically labeled so that the final plot has individual peaks that each correspond to a single amino acid in the protein. After conducting a series of experiments to assign each peak to its corresponding amino acid, the protein’s structure can finally be solved. Much like Cryo-EM, there is a size limitation to the samples that can be studied with NMR. Traditional solution-based NMR is ideal for studying smaller molecules, so unlike Cryo-EM, it is primarily used for obtaining structures of the individual subunits of spike protein rather than the fully assembled complex [6].
What Systems are Used?
By a wide margin, the most commonly used expression system for proteins of interest is the prokaryotic organism E. coli. This system is commonly used because of its fast growth, high yield, and reliability. However, for applications that rely on specific immune-complex interactions, such as ELISA assays or vaccines, prokaryotic systems are not ideal [7].
Furthermore, it is very difficult to express the receptor-binding domain in prokaryotic systems because these systems cannot carry out glycosylation processes that are important for the proper folding of that domain. For the produced spike protein to be used in clinical applications such as antigen testing or vaccine development, it must be expressed using a mammalian system. Of these mammalian expression systems, there are a myriad of choices, but the most commonly used cell line for this purpose is the HEK293 cell line, with Expi293 performing the best [7]. Apart from the specificity that is required from spike protein produced from mammalian cells, it is also important that these expression systems produce a protein that can form stable trimers to better represent the spike proteins found on the mature virus. While the recent COVID-19 pandemic has shifted most of the focus towards clinical applications, and mammalian expression systems along with them, traditional prokaryotic expression systems are still in use. E. coli expression systems are ideal for producing large amounts of stable spike protein subunits, save for the aforementioned receptor-binding domain [8]. These subunits can be used for structural characterization and can even be used to form complete trimers (provided all three subunits are expressed) that can be used for antigen-detection assays such as ELISAs (though not at the specificity required for the aforementioned clinical applications) [8]. For example, E. coli expression systems are used for the structural characterization of the receptor-binding domain of spike protein. Yeast expression systems can also be used to purify spike protein, though not in as large quantities as from E. coli. Yeast expression systems are still superior to mammalian systems in terms of yield and scalability, and their eukaryotic nature overcomes the glycosylation issues that prevent the receptor-binding domain from being synthesized in large quantities from prokaryotic cells [9]. This makes them a candidate choice for an alternate expression system to mammalian cell lines, though the difference in final protein structure from yeast expression compared to mammalian expression is still under investigation.
What Constructs?
While not as important as the type of organism in which to express spike protein, plasmid constructs can still be vital to the final yield and the functionality of the final product. For prokaryotic expression, standard subcloning approaches may be utilized to insert a PCR-amplified spike protein subunit gene into plasmid vectors. While the type of plasmid to use varies depending on the desired application and expression workflow, some examples from the literature include pET32a, pETGEX-4t-2, and pET-MBP [10]. Keep in mind, these expression vectors should not be used to amplify the gene sequence before subcloning. For that application, a vector such as pET-S would be more appropriate. The process for making constructs for mammalian systems is very similar, though different plasmid vectors must be used. For example, the mammalian plasmid vector pCDNA3.1 was used to express the receptor-binding domain of spike protein along with a poly-Histidine tag and two extraneous sequences tailor-made for that specific experiment [11]. This trend also holds true for expression in yeast, the subcloning process and desired constructs are more or less identical with the only major difference being the template vector that the gene of interest is cloned into, an example for yeast would be pPICZalpha. This flexibility associated with construct design has certainly aided research into spike protein, though challenges still exist in the purification process, which will be discussed in a later section. Regardless, it is important to consider the characteristics of each expression system before choosing a template vector to make the final construct.
What is the Current Interest?
Like any viral protein, most of the current interest centered on spike protein relates to clinical applications such as vaccines and antibody screenings. This is especially true after the COVID-19 pandemic, wherein spike protein became the target of vaccine-based research. Since coronavirus infections are always initiated by the binding of spike protein to the ACE2 receptor, targeting antibodies to this protein will prevent viral particles from starting new infections and help to target cells that are currently undergoing the process of infection. Since spike protein is subject to degradation by the innate immune system if injected directly, key vaccine strategies involve the production of spike protein mRNA to induce human cells to produce the protein on their own [10]. The purpose of this is to utilize the acquired immune system to produce antibodies that will recognize virus particles should they enter the body. While most previously discussed interest in spike protein has related to its structure, this key application also relies heavily on sequencing. While one of the biggest hurdles to any vaccine development is the delivery aspect, mRNA vaccines simply would not be possible without the fully sequenced spike protein gene from which to make mRNA. Given the immense amount of diversity between different types of coronaviruses, and the fact that the spike protein, in particular, is subject to rapid mutation, getting a useful gene sequence is not as simple as sequencing the already well-characterized SARS and MERS viruses. Fortunately, after the identification of SARS-CoV2 in late 2019, the virus was isolated and had its genome sequenced to provide a consensus sequence for the spike protein in early 2020. This spurred a further investigation into the comparison between the gene sequences between SARS-CoV2 and other human infecting coronaviruses to better ascertain the type of receptor that it binds to. Antibodies that are produced in this manner, as well as antibodies that are produced as part of a natural immune response to the virus, can be detected using a diagnostic test to assess whether a person has been previously infected with SARS-CoV2 or has produced antibodies against spike protein. Further interest in spike protein is centered on the aforementioned structural studies to better understand viral infection, subunit assembly, and receptor binding [11]. All of these factors are critical to understanding how coronaviruses are capable of infecting hosts and how they assemble in living cells. Additionally, the high rate of mutation among these viruses means that structures are subject to slight variations over time. While the structures of the membrane and nucleocapsid proteins are highly conserved, spike protein tends to accumulate slight changes that correspond with host-virus interactions. For example, SARS-CoV2 has already produced variants (namely, the Delta variant) that have shown to be more lethal and/or infectious than the original strain that originated from Wuhan, China in 2019. Since spike protein is responsible for starting viral infections, most of the differences between these variants likely lie in the structure of spike protein, as more efficient receptor binding will lead to a greater infection rate. Initial data on spike protein structure of the delta variant showed changes in the furin cleavage site (a proteolysis sequence essential to viral infection), the deletion of residues 156-157 of the N-terminal domain, and the point mutation of amino acid 158 to a glycine (N terminal mutations of this nature typically correspond to reduced detection from the immune system). While mutations in the other viral proteins can certainly lead to a more deadly virus, spike protein will remain the center of attention due to its clinical importance.
What are the Challenges to Purifying Spike Protein?
Despite the vast amount of attention that has been centered around spike protein over the past decade, there remain purification issues that hinder further research. The first is that pre-fusion spike protein requires post-translational modification to adopt its mature form [12]. Traditional prokaryotic expression systems are not capable of delivering the post-translational modification required to study the entire protein, so truncated forms that do not require this modification are used instead. This approach is sufficient when focusing on individual parts of the protein, such as the receptor-binding domain, but is inadequate for use in diagnostic assays or other applications that require a fully-assembled spike protein trimer. When spike protein is expressed in prokaryotic systems, the resulting protein forms inclusion bodies [13]. Obtaining soluble protein from these inclusion bodies involves aggressively denaturing the inclusion bodies with chaotropic chemicals before dialyzing these chemicals out to refold soluble spike protein monomers. This approach is not ideal as its yield is very low due to a subpopulation of protein that is irreversibly damaged by the aggressive denaturation step [13]. To produce full-length subunits, production must be conducted in a eukaryotic expression system that is capable of carrying out the post-translational modification required. The main drawback to this approach is that the yields from most well-characterized yeast and mammalian cell lines are very low when compared to prokaryotic expression. While it is possible to get high yields from eukaryotic expression systems, a standardized protocol is yet to be seen in the literature. Even when full-length subunits are produced, some challenges remain. Spike protein is found in two major conformations: “up” and “down”. Spike protein can only bind to a host receptor when found in the “up” conformation, thus making it the more physiologically relevant form. Unfortunately, spike proteins found in the “up” conformation are very unstable in solution, even when kept at four degrees Celsius [12]. While the “down” conformation is much more stable, it cannot be used for applications such as receptor binding assays. Further research into spike protein could be aided significantly by developing a method to stabilize the “up” conformation in solution. Spike protein trimers are also extremely difficult to purify due to their size (~670 kDa), leading many groups to purify the monomers and allow them to oligomerize after purification. While there are certainly many challenges associated with expressing and purifying spike protein, many of them have already been addressed with promising directions for future groups to take them.
What is Known About Its Structure?
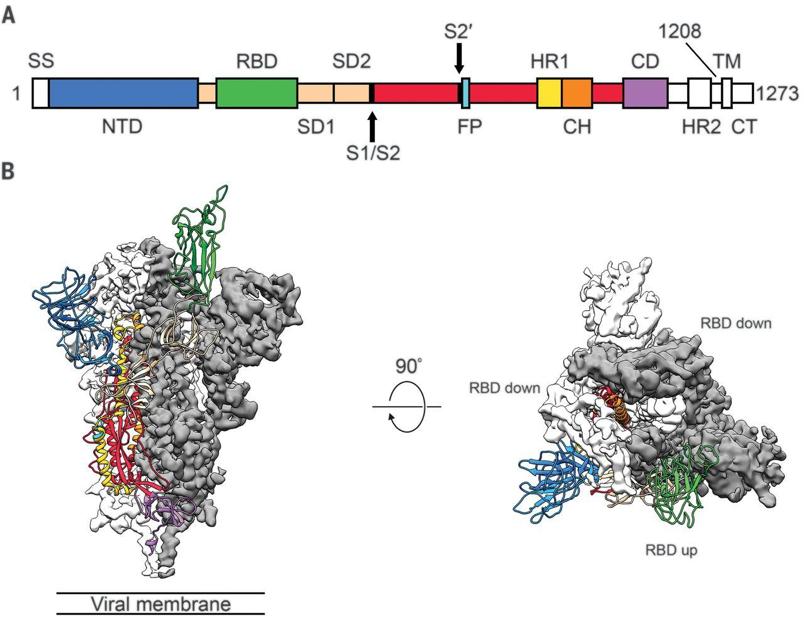
Individual spike protein monomers are composed of three major domains. These domains, in order of N to C terminal, are the N-terminal glycan binding, the ACE2 receptor binding, fusion peptide, and a heptad repeat coiled-coil [14]. Spike proteins can adopt two different conformations: one with the receptor-binding domain pointing up, and another with the receptor-binding domain pointing down. Coronaviruses can only bind a host cell with the spike protein in the up conformation, but this is less stable and makes it more vulnerable to immune system responses, so it spends most of its time in the down conformation [14]. The human enzyme angiotensin-converting enzyme 2 (ACE2) was identified as the receptor for the spike proteins of both SARS-CoV and SARS-CoV2, though MERS-CoV binds to dipeptidyl peptidase 4 (DPP4) [15]. Once fully assembled into the mature trimer, there are three distinct regions of interest: S1, S2, and IC. IC anchors the protein to the transmembrane region of the viral envelope (coronaviruses cloak themselves in lipid membranes that surround the protein-composed capsid). S1 is the crown-shaped region that contains the receptor-binding domain. Once a host cell is bound, S1 is proteolyzed which allows S2 to present a fusion peptide that further buries the virus into the host cell. Once this is achieved, the viral envelope merges with the cell membrane to complete the infection [15]. Spike protein is also decorated in glycosylation sites throughout the S1 and S2 regions. Attachment of sugars to the protein is theorized to help evade immune system responses.
Figure 1, from Wrapp et al. (2020) Science 367:1260. A) Sequence map of spike protein, with highlighted monomers. B) Cryo-EM structure of prefusion spike protein monomer with one RBD in the receptor-accessible “up” conformation.
Conclusion
Spike protein is a highly conserved trimeric protein that is essential for the survival of all coronaviruses. Despite knowledge of coronavirus infections dating back to the early 20th century, the many mysteries associated with this class of viruses are only beginning to be unraveled. Coronavirus research has taken off dramatically after the COVID-19 pandemic, leading to many developments in the structural characterization of spike protein as well as developing new ways to produce it in the lab. Structural characterization techniques such as NMR and Cryo-EM have revealed that spike protein has two primary conformations: a receptor-accessible “up” and receptor-inaccessible “down”. It is theorized that this sheathing of the receptor-binding domain helps to stabilize the protein for traveling long distances and shielding it from recognition by the host’s immune system. While many facets of spike protein structure and function have been elucidated, many challenges remain. An optimal expression system has yet to be determined, while there are benefits and downsides to all of the popular choices an ideal system has not been identified that can produce viable spike protein for all applications. Two applications of recent importance are serological assays and vaccine development, both of which are critical to clinical efforts against SARS-CoV2. Bacterial expression systems do not have the post-translational machinery necessary to produce full-length monomers, and mammalian expression systems are not high-throughput enough for the astronomically high demand imposed by this pandemic. While an impressive amount of progress has been achieved in just under two short years, there are still many challenges left to overcome.
References
- Kahn, J. S., & McIntosh, K. (2005). History and Recent Advances in Coronavirus Discovery. Pediatric Infectious Disease Journal, 24(11). https://doi.org/10.1097/01.inf.0000188166.17324.60
- Belouzard, S., Millet, J. K., Licitra, B. N., & Whittaker, G. R. (2012). Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, Vol. 4, Pages 1011-1033, 4(6), 1011–1033. https://doi.org/10.3390/V4061011
- Du, L., He, Y., Zhou, Y., Liu, S., Zheng, B.-J., & Jiang, S. (2009). The spike protein of SARS-CoV — a target for vaccine and therapeutic development. Nature Reviews Microbiology 2009 7:3, 7(3), 226–236. https://doi.org/10.1038/nrmicro2090
- Kirchdoerfer, R. N., Cottrell, C. A., Wang, N., Pallesen, J., Yassine, H. M., Turner, H. L., Corbett, K. S., Graham, B. S., McLellan, J. S., & Ward, A. B. (2016). Pre-fusion structure of a human coronavirus spike protein. Nature, 531(7592). https://doi.org/10.1038/nature17200
- Bai, X. chen, McMullan, G., & Scheres, S. H. W. (2015). How cryo-EM is revolutionizing structural biology. In Trends in Biochemical Sciences (Vol. 40, Issue 1). https://doi.org/10.1016/j.tibs.2014.10.005
- Mahajan, M., Chatterjee, D., Bhuvaneswari, K., Pillay, S., & Bhattacharjya, S. (2018). NMR structure and localization of a large fragment of the SARS-CoV fusion protein: Implications in viral cell fusion. Biochimica et Biophysica Acta – Biomembranes, 1860(2). https://doi.org/10.1016/j.bbamem.2017.10.002
- Esposito, D., Mehalko, J., Drew, M., Snead, K., Wall, V., Taylor, T., Frank, P., Denson, J.-P., Hong, M., Gulten, G., Sadtler, K., Messing, S., & Gillette, W. (2020). Optimizing high-yield production of SARS-CoV-2 soluble spike trimers for serology assays. Protein Expression and Purification, 174. https://doi.org/10.1016/j.pep.2020.105686
- Chen, J., Miao, L., Li, J.-M., Li, Y.-Y., Zhu, Q.-Y., Zhou, C.-L., Fang, H.-Q., & Chen, H.-P. (2005). Receptor-binding domain of SARS-Cov spike protein: Soluble expression in E.coli, purification and functional characterization. World Journal of Gastroenterology : WJG, 11(39), 6159. https://doi.org/10.3748/WJG.V11.I39.6159
- Consortium, A. A. (2020). Structural and functional comparison of SARS-CoV-2-spike receptor binding domain produced in Pichia pastoris and mammalian cells. Scientific Reports 2020 10:1, 10(1), 1–18. https://doi.org/10.1038/s41598-020-78711-6
- Malik, J. A., Mulla, A. H., Farooqi, T., Pottoo, F. H., Anwar, S., & Rengasamy, K. R. R. (2021). Targets and strategies for vaccine development against SARS-CoV-2. Biomedicine & Pharmacotherapy, 137, 111254. https://doi.org/10.1016/J.BIOPHA.2021.111254
- Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G. F., & Tan, W. (2020). A Novel Coronavirus from Patients with Pneumonia in China, 2019. Https://Doi.Org/10.1056/NEJMoa2001017, 382(8), 727–733. https://doi.org/10.1056/NEJMOA2001017
- Edwards, R. J., Mansouri, K., Stalls, V., Manne, K., Watts, B., Parks, R., Janowska, K., Gobeil, S. M. C., Kopp, M., Li, D., Lu, X., Mu, Z., Deyton, M., Oguin, T. H., Sprenz, J., Williams, W., Saunders, K. O., Montefiori, D., Sempowski, G. D., … Acharya, P. (2021). Cold sensitivity of the SARS-CoV-2 spike ectodomain. Nature Structural and Molecular Biology, 28(2), 128–131. https://doi.org/10.1038/s41594-020-00547-5
- Piao, D. C., Lee, Y. S., Bok, J. D., Cho, C. S., Hong, Z. S., Kang, S. K., & Choi, Y. J. (2016). Production of soluble truncated spike protein of porcine epidemic diarrhea virus from inclusion bodies of Escherichia coli through refolding. Protein Expression and Purification, 126, 77–83. https://doi.org/10.1016/j.pep.2016.05.018
- Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C.-L., Abiona, O., Graham, B. S., & McLellan, J. S. (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science, 367(6483). https://doi.org/10.1126/science.abb2507
- Walls, A. C., Park, Y.-J., Tortorici, M. A., Wall, A., McGuire, A. T., & Veesler, D. (2020). Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell, 181(2). https://doi.org/10.1016/j.cell.2020.02.058
